
Ramsey A, Mallouk A, Sharma A, Baxter T. Timeline in pictures of Oral Aphthae as a presenting symptom fo Behcet’s Disease. HPHR. 2022;67.
High prevalence of BD (420 per 100,000) has been reported in Turkey 1 with lowest prevalence of 0.38 per 100,000 being reported in North America 2. In sub‐Saharan Africa, the prevalence of BD is not known but few cases have been reported elsewhere 4-6, with only one case being reported from Tanzania over 40 years ago 7.
We are reporting a 60‐year‐old Turkish male who presented to our outpatient clinic frustrated by multiple visits to many doctors and clinics for his seemingly puzzling symptoms. The patient initially had blisters all over his lips and tongue. He went to a walk-in clinic and was told he had Herpes. The patient also had a blister on his penis. However, the patient denied being sexually active for years which prompted us to conduct further investigation and search of the literature. Two weeks later, the patient stated that the ulcers were healing and everything was beginning to look normal again. The next day the patient woke up with back pain radiating to the right side of the mid-thoracic area for which he went to the ED. He was diagnosed with a kidney stone on CT imaging. The patient described 2 similar episodes of oral ulcerations over the previous year. The mouth ulcers started gradually in the buccal cavity, tongue, and lips. There have been periods of complete healing and recurrences. His recurrences were neither bleeding nor discharging. He denied history of epigastric pain, painful defecation, painful micturition, hematuria, reduced amount of urine, or any history suggestive of sexually transmitted diseases in the past. There was no blurred vision or photophobia. Over the course of his illness, the patient had neither fever nor weight loss.
He had no known history of allergy and had never been transfused with blood or blood products. All of his family members were healthy and none had similar illness. He is a former smoker and no history of drinking alcohol.
Physical examination revealed an anxious patient with multiple concerns about his health who was fully alert, cooperative, and afebrile. He had no oro-genital ulcerations, eye lesions or skin abnormalities at the time of the exam. There was no lymphadenopathy. Eye examination revealed normal visual acuity, normal visual fields, normal optic nerves and no signs of uveitis.
The blood pressure was 122/82 mmHg, the pulse rate was 68 beats per minute regular, the respiratory rate was 17 cycles per minutes, and the oxygen saturation was 96% in room air. Urogenital system examination revealed normal male genitalia. The physical examination of the rest of the systems was essentially normal.
The results from laboratory analysis done were complete blood count (hemoglobin level of 14.6 g/dL and a mean corpuscular volume 93.7 fl., all blood cell counts were within normal ranges), renal function test (normal range), fasting blood glucose (92MG/DL), and Pathergy test (negative). HLA-B27 antigen negative. The Erythrocyte Sedimentation Rate (ESR) was 18MM/HR and C-Reactive Protein (CRP) was 1.03mg/L. Herpes Simplex Virus (HSV) Type 1&2 were undetected in the serum. Varicella-Zoster Virus (VZV) PCR was negative. The diagnosis of BD was made according to the ISGBD 9 based on the presence of recurrent oral aphthae (≥3 times in 1 year) together with recurrent genital aphthae and the patient’s self-reported skin lesions.
The patient currently requires no treatment since his disease course seems to be in remission. Close monitoring of the various systemic symptoms of BD are advised and appropriate follow-up is recommended. Generally, BD responds well to steroids, with the combination of corticosteroids and immunosuppressant drugs being indicated when vital organs are involved 10.
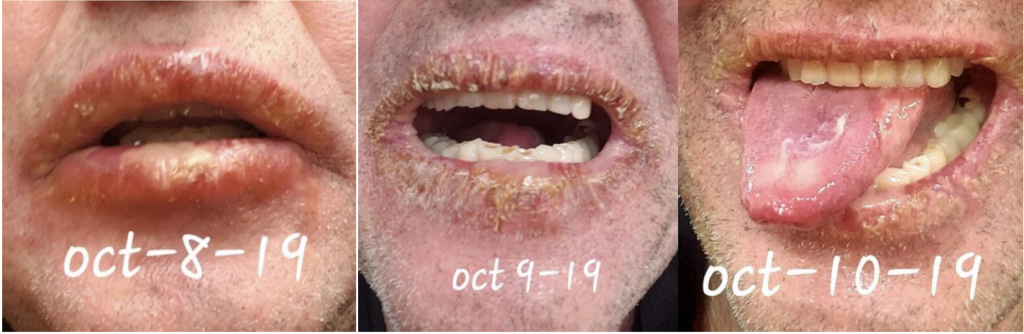
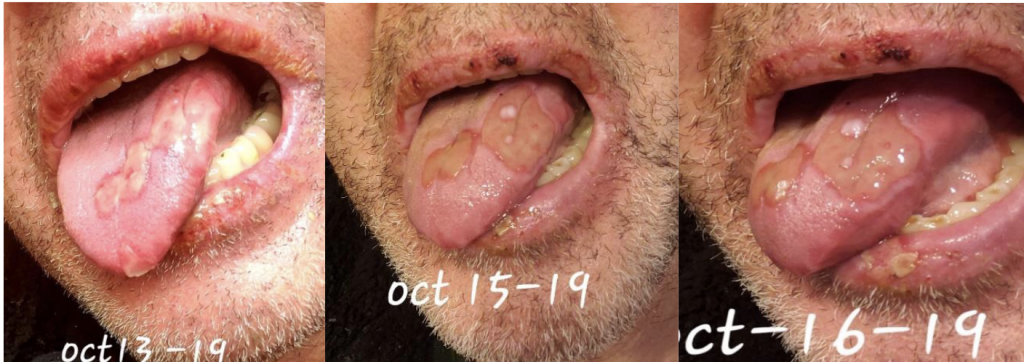
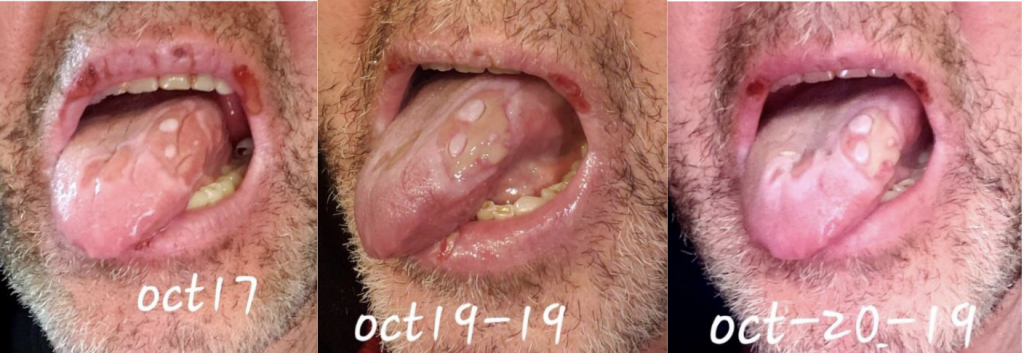
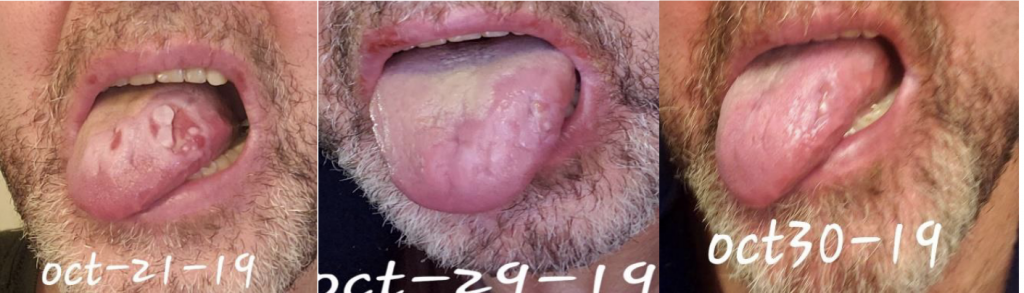
This case emphasizes the need to increase awareness among clinicians to recognize the various manifestations of BD so as to timely diagnose and offer prompt treatment. Further studies are needed to ascertain the prevalence and distribution of BD in the US as well as associated genetic factors.
The authors are thankful to the Family Medicine Residency Program Director at Arnot Ogden Medical Center and staff members at the Eastside Family Medicine clinic in St. Joeseph’s Medical Center in Elmira, New York for their support.
1 Yurdakul, S., V. Hamuryudan, and H. Yazici. 2004. Behcet syndrome. Curr. Opin. Rheumatol. 16: 38– 42
2 Calamia, K. T., F. C. Wilson, M. Icen, C. S. Crowson, S. E. Gabriel, and H. M. Kremers. 2009. Epidemiology and clinical characteristics of Behcet’s disease in the US: a population‐based study. Arthritis Rheum. 61: 600– 604.
3 de Menthon, M., M. P. Lavalley, C. Maldini, L. Guillevin, and A. Mahr. 2009. HLA‐B51/B5 and the risk of Behcet’s disease: a systematic review and meta‐analysis of case‐control genetic association studies. Arthritis Rheum. 61: 1287– 1296.
4 Haile, A. 1997. Behcets’ disease: a case report. Ethiop. Med. J. 35: 191– 199.
5 Verity, D. H., J. E. Marr, S. Ohno, G. R. Wallace, and M. R. Stanford. 1999. Behcet’s disease, the Silk Road and HLA‐B51: historical and geographical perspectives. Tissue Antigens 54: 213– 220.
6 Liozon, E., C. Roussin, X. Puechal, A. Garou, P. Valadier, I. Perinet, et al. 2011. Behcet’s disease in East African patients may not be unusual and is an HLA‐B51 negative condition: a case series from Mayotte (Comoros). Joint Bone Spine 78: 166– 170.
7 Makene, W. J. 1969. Behcet’s syndrome with central nervous system involvement in a Tanzanian African. East Afr. Med. J. 46: 199– 203.
8 Fietta, P. 2005. Behcet’s disease: familial clustering and immunogenetics. Clin. Exp. Rheumatol. 23( 4 Suppl. 38): S96– S105.
9 International Study Group for Behcet’s Disease. 1990. Criteria for diagnosis of Behcet’s disease. International study group for Behcet’s Disease. Lancet 335: 1078– 1080.
10 Kaklamani, V. G., and P. G. Kaklamanis. 2001. Treatment of Behcet’s disease–an update. Semin. Arthritis Rheum. 30: 299– 312.
11 J Eur Acad Dermatol Venereol. 2014 Mar;28(3):338-47. doi: 10.1111/jdv.12107. Epub 2013 Feb 26
HPHR.org was designed by ComputerAlly.com.
Visit HPHR’s publisher, the Boston Congress of Public Health (BCPH).
Email communications@bcph.org for more information.
Click below to make a tax-deductible donation supporting the educational initiatives of the Boston Congress of Public Health, publisher of HPHR Journal.![]()